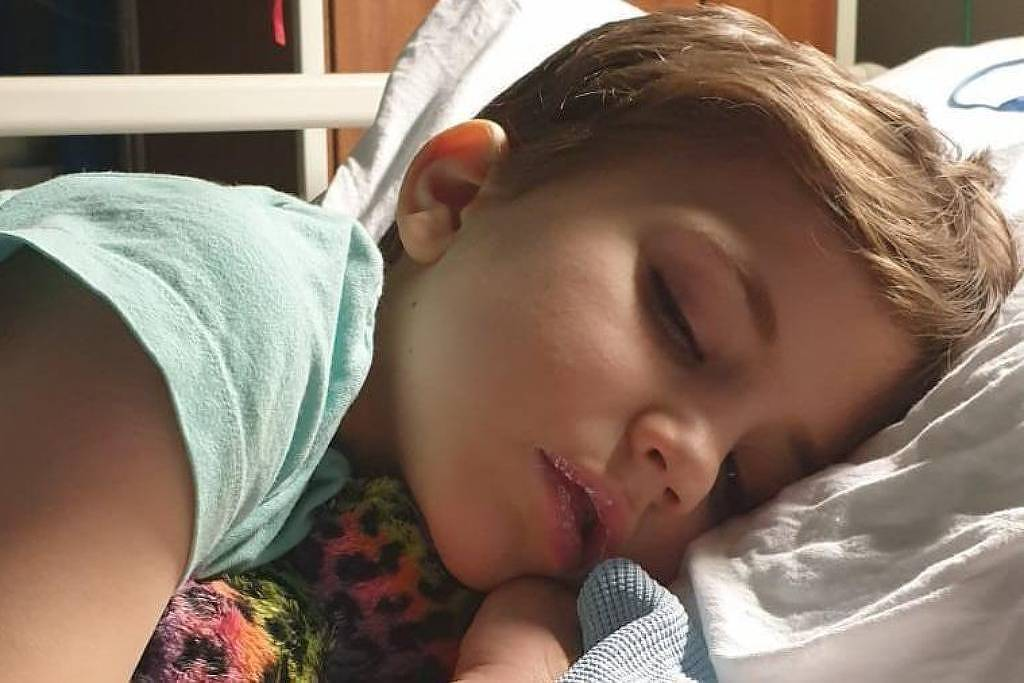
O caso do menino Roo, de 7 anos, revelou um risco pouco discutido na cadeia de produção e fiscalização de suplementos. Internado após perda de peso, sede intensa e vômitos, ele foi diagnosticado com lesão renal aguda e hipercalcemia. Exames e investigações apontaram que o frasco de vitamina D3 em gotas prescrito tinha concentração cerca de sete vezes maior do que o padrão — um defeito presente em um de dois lotes distribuídos no Reino Unido.
A prescrição, feita em dezembro de 2024 para um regime de 12 semanas com dose elevada, havia sido cumprida em cerca de dois terços quando Roo piorou. Equipes do Hospital Crosshouse, em Kilmarnock, e do Hospital Real Infantil de Glasgow acompanharam o caso; um especialista disse à BBC que a criança teria morrido caso o tratamento tivesse sido levado até o fim. O episódio deixou claro o efeito tóxico da hipervitaminose D quando associada a produtos mal dosados.
O acidente também expõe uma lacuna regulatória no Reino Unido: doses mais altas de vitamina D, apesar de receitadas por médicos, ainda são tratadas como suplemento alimentar e não reguladas pela MHRA, que atua sobre medicamentos. A Agência de Padrões Alimentares (FSA) é a responsável por suplementos, e a MHRA afirma cooperar com a FSA. Especialistas consultados pela imprensa defendem revisão da classificação e maior supervisão sobre lotes e concentrações.
No Brasil, a Anvisa fiscaliza tanto medicamentos quanto suplementos, uma diferença institucional que evita, em tese, o mesmo vácuo regulatório. O caso de Roo, porém, evidencia consequências concretas: risco de morte, dúvidas sobre controles de qualidade e necessidade de respostas rápidas de órgãos e fabricantes. Para evitar novos episódios, o debate agora tende a pressionar mudanças na regulação, transparência sobre lotes defeituosos e procedimentos de comunicação ao público.